Differential expression analysis of cancer data
May 31, 2024
Abstract
Here we perform a differential expression analysis of the filtered and normalized RNA-seq expression profiles of cancer data.
1 Importing processed and filtered data
We start by importing the previously filtered and normalized RNA-seq data.
library(SummarizedExperiment)
library(edgeR)
dgeR.filt <- readRDS(file.path("_processed_data", "dgeR.filt.rds"))
seR.filt <- readRDS(file.path("_processed_data", "seR.filt.rds"))
dgeB.filt <- readRDS(file.path("_processed_data", "dgeB.filt.rds"))
seB.filt <- readRDS(file.path("_processed_data", "seB.filt.rds"))
dgeR.filt.training <- readRDS(file.path("_processed_data",
"dgeR.filt.training.rds"))
seR.filt.training <- readRDS(file.path("_processed_data",
"seR.filt.training.rds"))
dgeB.filt.testing <- readRDS(file.path("_processed_data",
"dgeB.filt.testing.rds"))
seB.filt.testing <- readRDS(file.path("_processed_data",
"seB.filt.testing.rds"))2 Roskams et al. (2022) data
Design matrices.
mod <- model.matrix(~ Tumor + Sex, colData(seR.filt.training))
mod0 <- model.matrix(~ Sex, colData(seR.filt.training))Estimate surrogate variables (SVs).
library(sva)
IQRs <- apply(assays(seR.filt.training)$logCPM, 1, IQR)
mask <- IQRs > quantile(IQRs, prob=0.9)
sv <- sva(assays(seR.filt.training)$logCPM[mask, ],
mod=mod, mod0=mod0)
Number of significant surrogate variables is: 3
Iteration (out of 5 ):1 2 3 4 5 mod <- cbind(mod, sv$sv)Fit linear models using voom (Law et al. 2014) with sample weights. Figure 1 shows the mean-variance trend of the data.
First sample weights (min/max) 0.2191654/1.9738444
Final sample weights (min/max) 0.2301639/1.9165526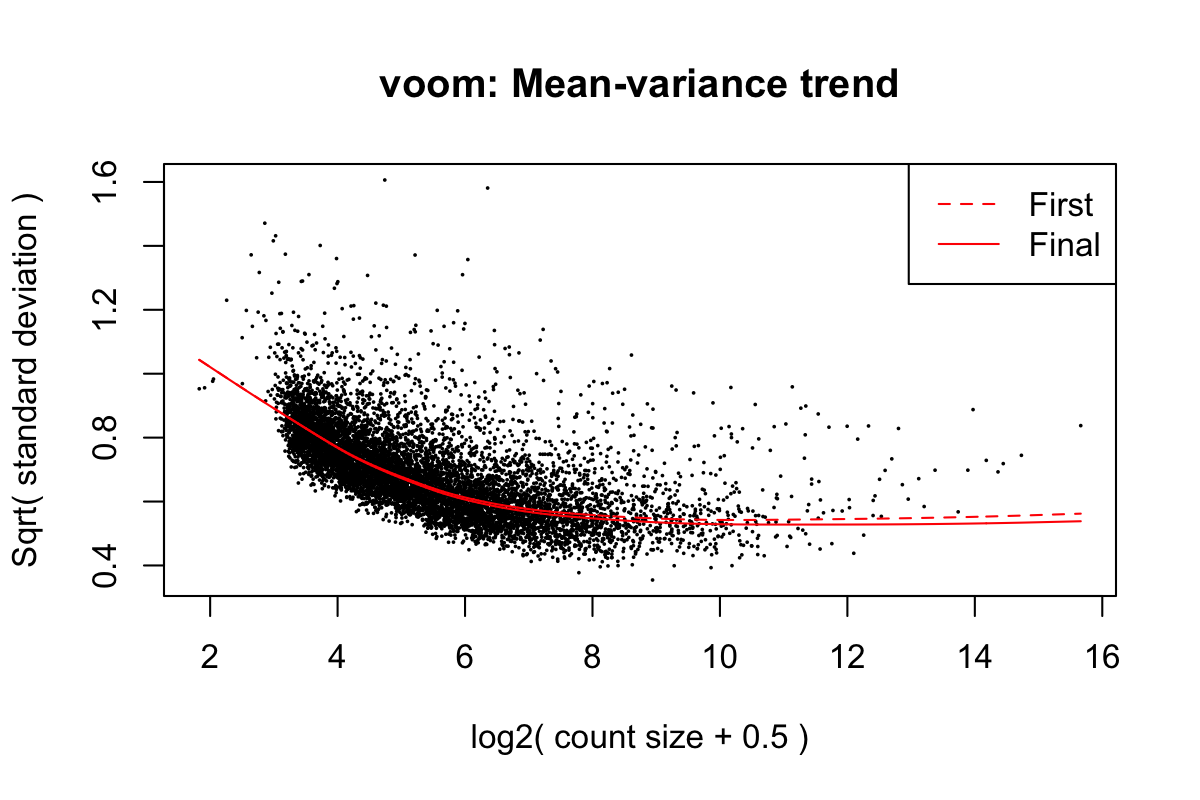
Figure 1: Mean-variance trend in the training data from Roskams et al. (2022)
Calculate moderated t-statistics using an empirical Bayes procedure and examine the extent of differential expression (DE) with an FDR < 5%.
fit.training <- eBayes(fit.training, robust=TRUE)
res.training <- decideTests(fit.training, p.value=0.05)
summary(res.training)
(Intercept) Tumoryes Sexmale
Down 0 435 0 4079 11 1011
NotSig 33 7775 8380 1405 8367 6260
Up 8354 177 7 2903 9 1116Fetch summary DE statistics for all genes.
tt.training <- topTable(fit.training, coef="Tumoryes", n=Inf,
sort.by="p")Display the p-value distribution in Figure 2.
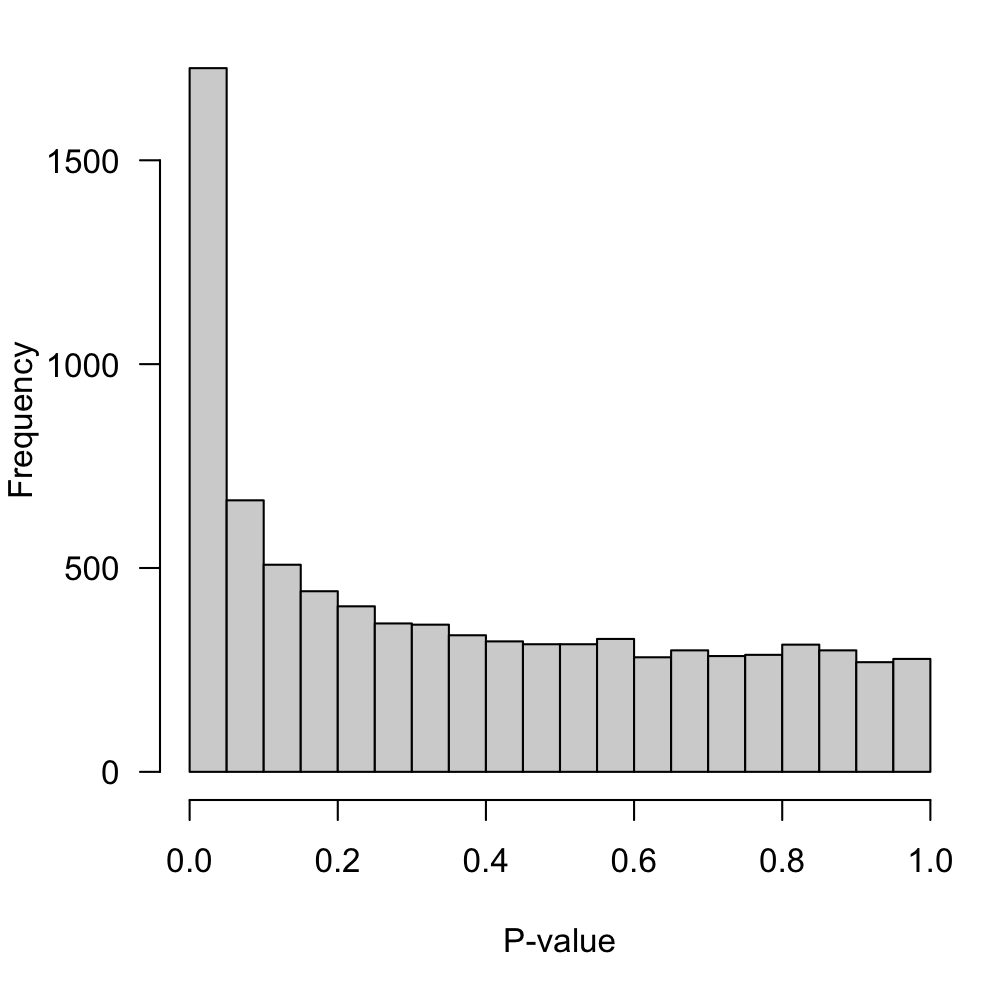
Figure 2: P-value distribution for the null hypothesis of no-DE
Select DE genes with FDR < 5%.
mask <- tt.training$adj.P.Val < 0.05
DEgenes.training <- rownames(tt.training)[mask]
length(DEgenes.training)
[1] 612Display volcano and MA plots in Figure 3.
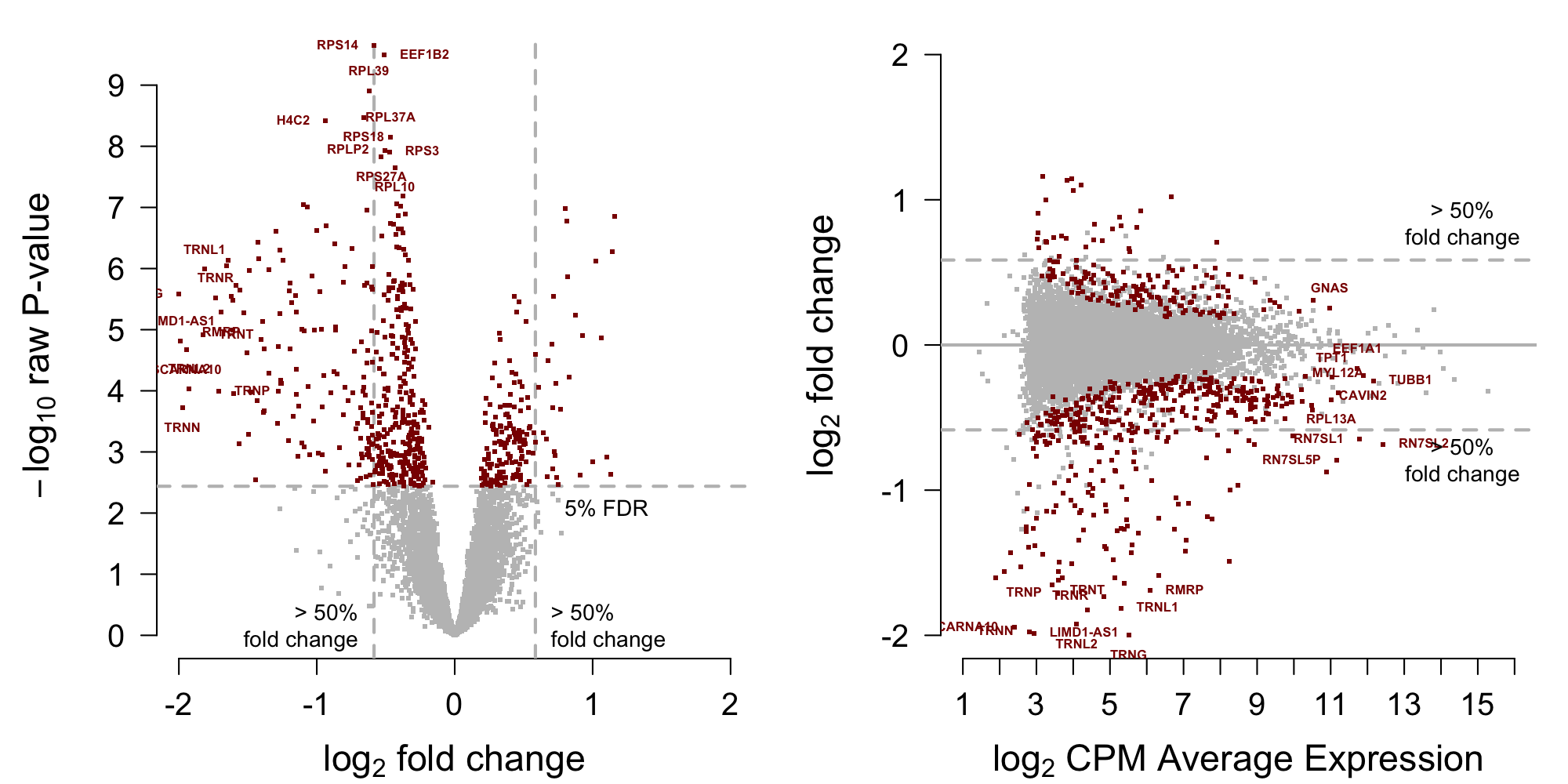
Figure 3: MA-plot training data from Roskams et al. (2022)
saveRDS(DEgenes.training, file=file.path("_processed_data", "DEgenes.trainingR.rds"))3 Block et al. (2022) data
Design matrices.
mod <- model.matrix(~ Tumor + Sex, colData(seB.filt.testing))
mod0 <- model.matrix(~ Sex, colData(seB.filt.testing))Estimate surrogate variables (SVs).
library(sva)
IQRs <- apply(assays(seB.filt.testing)$logCPM, 1, IQR)
mask <- IQRs > quantile(IQRs, prob=0.9)
sv <- sva(assays(seB.filt.testing)$logCPM[mask, ],
mod=mod, mod0=mod0)
Number of significant surrogate variables is: 2
Iteration (out of 5 ):1 2 3 4 5 mod <- cbind(mod, sv$sv)Fit linear models using voom (Law et al. 2014) with sample weights. Figure 1 shows the mean-variance trend of the data.
First sample weights (min/max) 0.07543019/3.55468768
Final sample weights (min/max) 0.0952618/3.8255265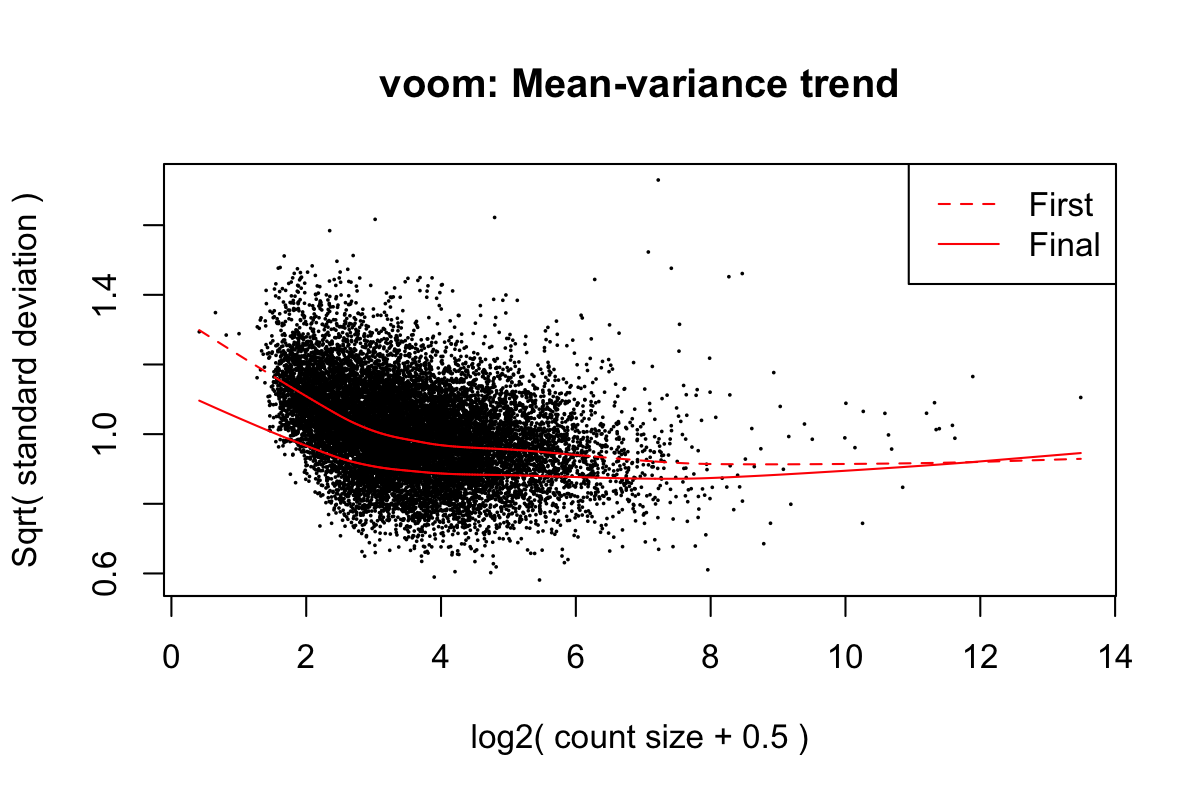
Figure 4: Mean-variance trend in the testing data from Block et al. (2022)
Calculate moderated t-statistics using an empirical Bayes procedure and examine the extent of differential expression (DE) with an FDR < 5%.
fit.testing <- eBayes(fit.testing, robust=TRUE)
res.testing <- decideTests(fit.testing, p.value=0.05)
summary(res.testing)
(Intercept) Tumoryes Sexmale
Down 0 101 670 7473 6452
NotSig 4 15463 14676 4012 7633
Up 15609 49 267 4128 1528Fetch summary DE statistics for all genes.
tt.testing <- topTable(fit.testing, coef="Tumoryes", n=Inf,
sort.by="p")Display the p-value distribution in Figure 5.
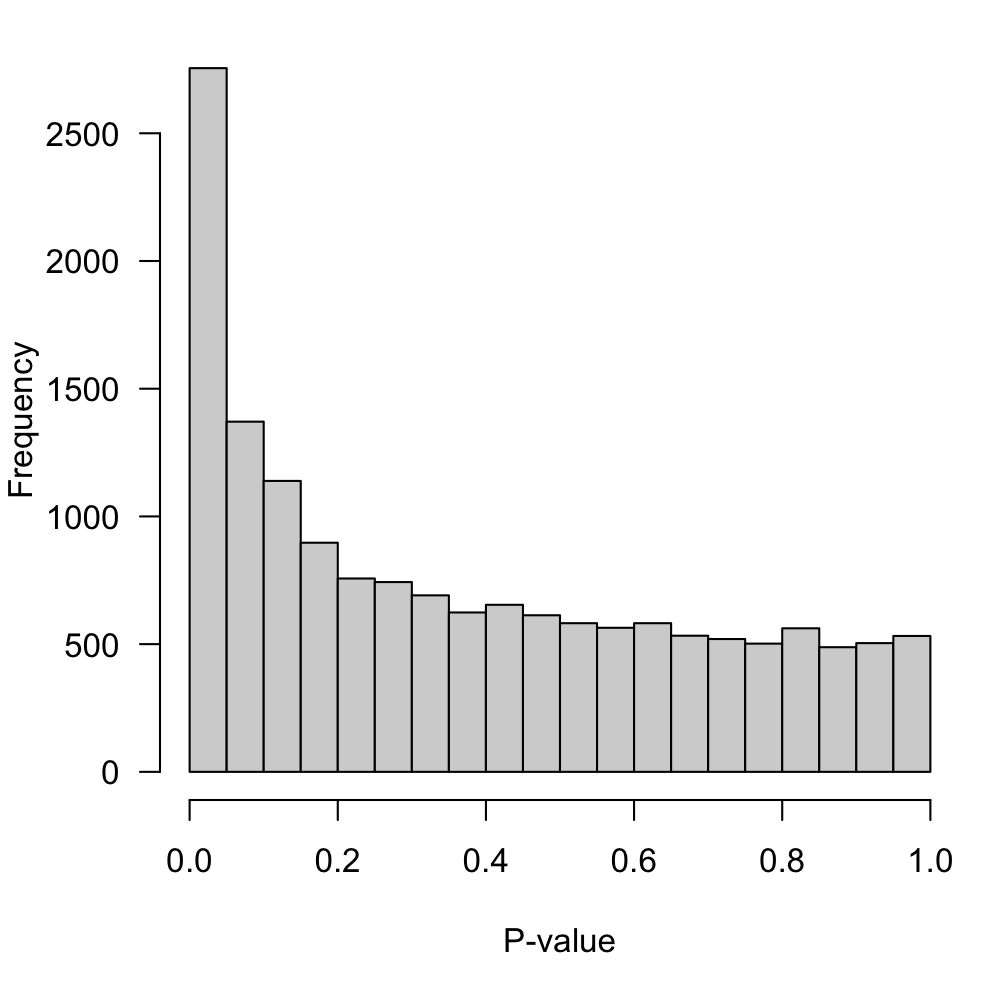
Figure 5: P-value distribution for the null hypothesis of no-DE
Select DE genes with FDR < 5%.
mask <- tt.testing$adj.P.Val < 0.05
DEgenes.testing <- rownames(tt.testing)[mask]
length(DEgenes.testing)
[1] 150Display MA-plot in Figure 6.
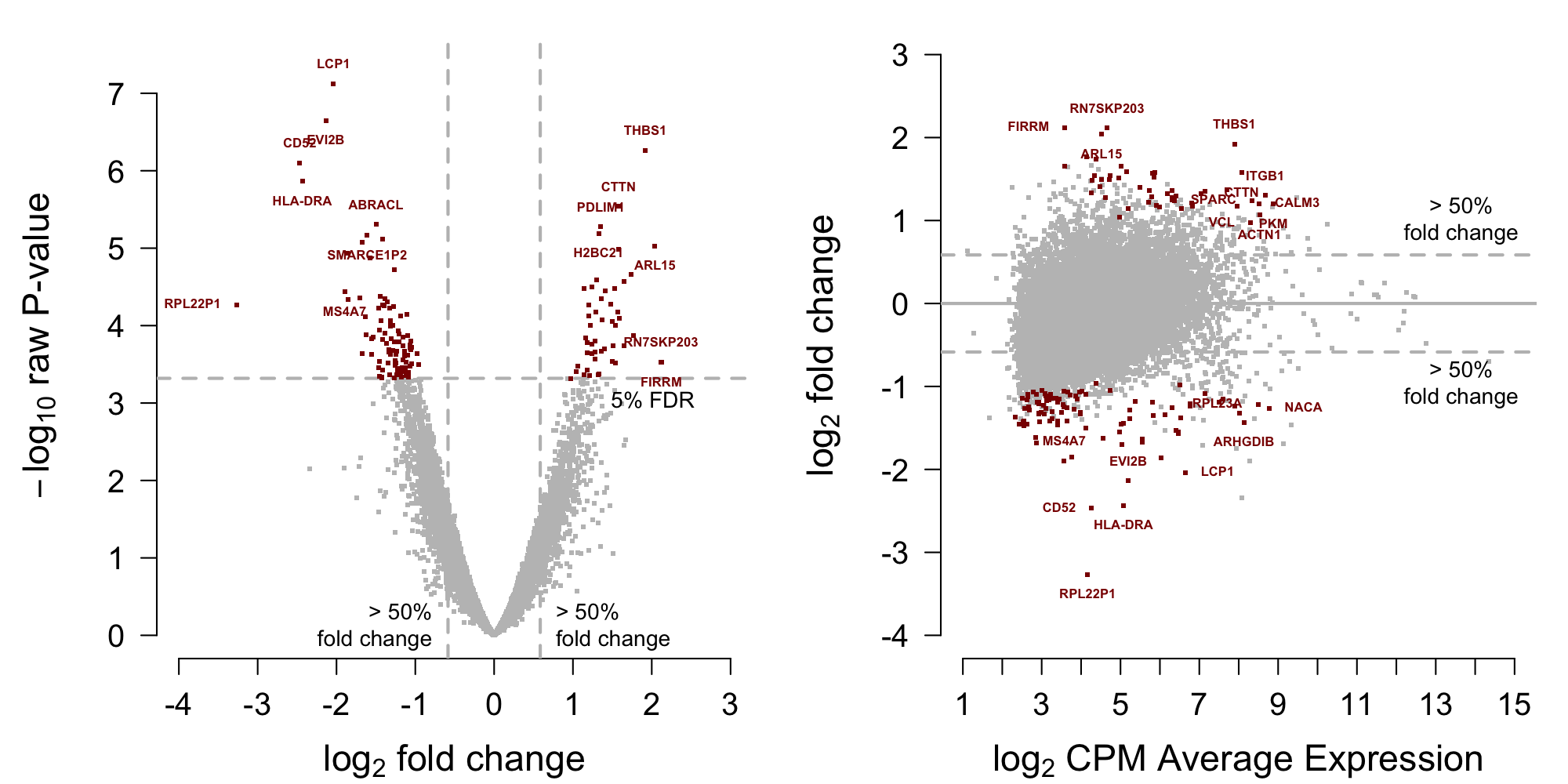
Figure 6: MA-plot testing data from Block et al. (2022)
4 Comparison of DE genes across datasets
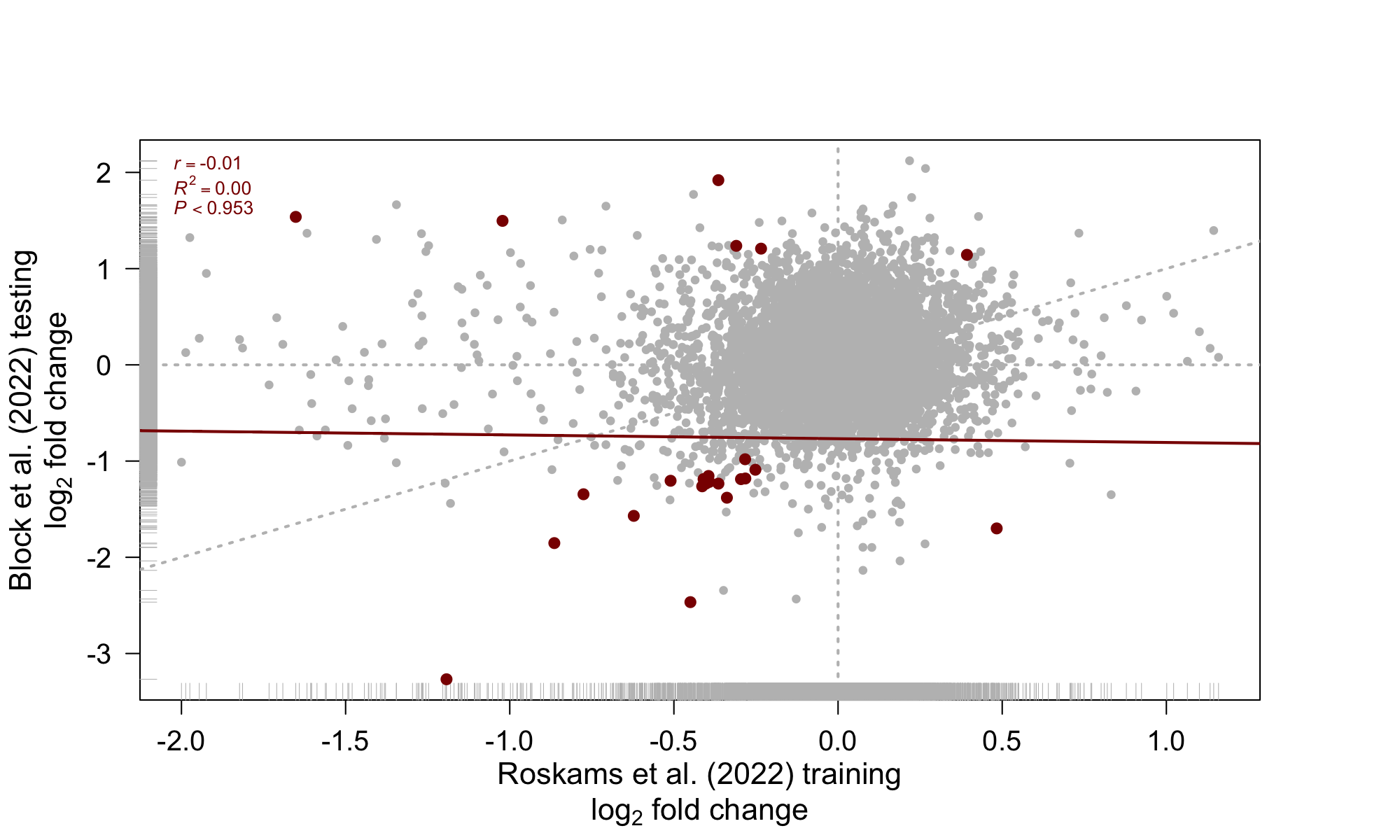
Figure 7: Comparison DE analyis across datasets
mask <- rownames(assay(seR.filt.training)) %in% commonDEgenes
commonDEgenes.name <- rowData(seR.filt.training)$Symbol[mask]
commonDEgenes.name
[1] "CD52" "MNDA" "RPL22" "RPL7P9" "CERS2" "RN7SL674P"
[7] "EEF1B2" "TUBA4A" "RN7SL752P" "RPL23AP42" "RPL22P1" "RPL34"
[13] "RPS23" "GIMAP7" "RPS20" "RPS13" "PFDN5" "NACA"
[19] "RPL21" "RPL36AL" "THBS1" "NUTF2" "RPL23A" "RPL39" 5 Session information
sessionInfo()
R version 4.4.0 (2024-04-24)
Platform: x86_64-apple-darwin20
Running under: macOS Ventura 13.5.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Europe/Madrid
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] plotrix_3.8-4 sva_3.52.0
[3] BiocParallel_1.38.0 genefilter_1.86.0
[5] mgcv_1.9-1 nlme_3.1-164
[7] edgeR_4.2.0 limma_3.60.2
[9] SummarizedExperiment_1.34.0 Biobase_2.64.0
[11] GenomicRanges_1.56.0 GenomeInfoDb_1.40.0
[13] IRanges_2.38.0 S4Vectors_0.42.0
[15] BiocGenerics_0.50.0 MatrixGenerics_1.16.0
[17] matrixStats_1.3.0 kableExtra_1.4.0
[19] knitr_1.46 BiocStyle_2.32.0
loaded via a namespace (and not attached):
[1] viridisLite_0.4.2 blob_1.2.4 Biostrings_2.72.0
[4] fastmap_1.2.0 XML_3.99-0.16.1 digest_0.6.35
[7] lifecycle_1.0.4 survival_3.5-8 statmod_1.5.0
[10] KEGGREST_1.44.0 RSQLite_2.3.6 magrittr_2.0.3
[13] compiler_4.4.0 rlang_1.1.3 sass_0.4.9
[16] tools_4.4.0 yaml_2.3.8 S4Arrays_1.4.1
[19] bit_4.0.5 DelayedArray_0.30.1 xml2_1.3.6
[22] abind_1.4-5 grid_4.4.0 xtable_1.8-4
[25] colorspace_2.1-0 scales_1.3.0 tinytex_0.51
[28] cli_3.6.2 rmarkdown_2.27 crayon_1.5.2
[31] rstudioapi_0.16.0 httr_1.4.7 DBI_1.2.2
[34] cachem_1.1.0 stringr_1.5.1 zlibbioc_1.50.0
[37] splines_4.4.0 parallel_4.4.0 AnnotationDbi_1.66.0
[40] BiocManager_1.30.23 XVector_0.44.0 vctrs_0.6.5
[43] Matrix_1.7-0 jsonlite_1.8.8 bookdown_0.39
[46] bit64_4.0.5 systemfonts_1.1.0 locfit_1.5-9.9
[49] jquerylib_0.1.4 annotate_1.82.0 glue_1.7.0
[52] codetools_0.2-20 stringi_1.8.4 UCSC.utils_1.0.0
[55] munsell_0.5.1 htmltools_0.5.8.1 GenomeInfoDbData_1.2.12
[58] R6_2.5.1 evaluate_0.23 lattice_0.22-6
[61] highr_0.10 png_0.1-8 memoise_2.0.1
[64] bslib_0.7.0 Rcpp_1.0.12 svglite_2.1.3
[67] SparseArray_1.4.3 xfun_0.44